Research Sequencing Service: Supported Analysis Pipelines
Whole Genome/Targeted Sequencing
Whole genome and targeted sequencing are the main usage modality for modern Next Generation Sequencing platforms. Common applications include genotyping of organisms and tumor mutational profiling. The RSS has implemented the nf-core/Sarek bioinformatic pipeline. Sarek is based on the Broad Institutes best practices guidelines and is designed to detect germline or somatic variants on whole genome or targeted sequencing data. It supports any species with a reference genome, as well as can also handles tumor/normal pairs. More information can be found on the Sarek website.
Bulk RNA-Seq
RNA sequencing (RNA-Seq) is a highly sensitive and accurate sequencing approach for measuring gene and isoform expression across the transcriptome. Bulk RNA-Seq offers a number of advantages over gene expression arrays and RT-qPCR including a broad dynamic range that enables sensitive and accurate measurement of gene expression, no prior knowledge of gene expression profiles or open reading frames, and being applicable to any species regardless of the availability of a reference genome. The RSS has implemented the nf-core/rnaseq bioinformatic pipeline which can be used to analyze bulk RNA-Seq data obtained from organisms with a reference genome and associated annotation. More information can be found on the nf-core/rnaseq website.
10X Genomics Single-Cell Assays
Why Perform Single Cell Analysis?
Short-read sequencing technologies, such as the Illumina platform, allow for the digital tabulation of many individual DNA/cDNA fragments in parallel, thus offering the unique ability to detect low-level nucleic acid species within heterogeneous mixtures and facilitating the systematic investigation of cellular heterogeneity in DNA sequence, epigenetic state, and RNA expression levels. However, bulk analysis of nucleic acids necessarily decouples cell-specific information between any two reads. For example, information allowing for the phasing of detected genomic variants or information about gene expression levels in specific cell types cannot reliably be inferred from bulk measurements. The only solution to this issue is to sequence individual cells, which allows for the unambiguous assignment of data to specific cells.
Platform and Principle
The 10X Genomics Chromium X is a microfluidic reagent delivery system that partitions cells or nuclei and prepares parallel sequencing libraries such that all DNA/cDNA produced within a single-cell partition is derived from the same cell using a cell-specific sequencing barcode (Fig. 1B). In this way, barcoded fragments can be processed in bulk downstream and still be linked to the originating individual cell. The DLMP Chromium X controller can process between 100 and 730,000 single-cells in one run, depending on assay configuration, in a simple workflow (Fig. 1B). After sequencing, the raw data is parsed such that all cell-specific data are grouped, aligned, and analyzed. The data can then be visualized using the 10X Genomics Cell Ranger software. This platform supports single-cell gene expression, single-cell immune profiling, single-cell ATAC-Seq, and single-cell multi-ome combining ATAC and gene expression.
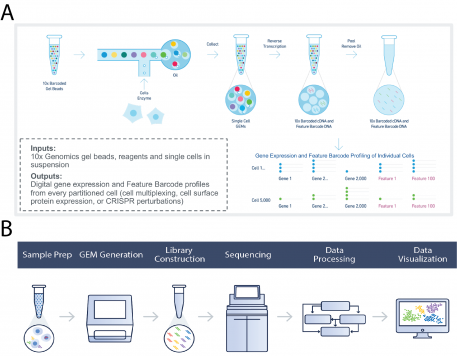
See a detailed description of flowchart A and B. Source: 10X Genomics Chromium product brochure.
Getting Started
Single-cell experiments can be performed on cell lines, disaggregated cells from tissues, and isolated nuclei. While highly versatile, single-cell sequencing experiments are complicated and typically require careful sample preparation and handling. We strongly recommend viewing the virtual training sessions that discusses best practices (UW NetID required). Stand alone slides of the presentation are also available. In addition, we require an initial hands-on training prior to using the instrument for the first time. Please contact us to schedule a consultation and training session. We will discuss your project goals, sample type, and estimated cost. The major determinants of overall project cost will be the number of cells, assay type, and sequencing requirements.
Duplex Sequencing (Duplex-Seq)
Why Perform Duplex-Seq?
Duplex-Seq is a technology used for high accuracy detection of ultra-low frequency mutations and is idea for studying somatic mutagenesis, tumor heterogeneity, and ultra-rare mutation detection. In theory, conventional NGS methods should be able to detect DNA subpopulations of any size by assessing a sufficient number of molecules. However, NGS platforms still generate errors at a substantial rate. Mistakes arising during DNA preparation, amplification, cluster generation, and sequencing itself, typically results in an error rate ~1%. In contrast, the vast majority of de novo somatic mutations are typically found to be several orders of magnitude lower than this background, making it impossible to study these low frequency mutations.
The Principal Behind Duplex-Seq
Developed at UW, Duplex-Seq is a consensus-based error correction method that uses double-stranded unique molecular identifiers (UMIs) to computationally obtain redundant sequence information from both strands of a DNA molecule. This method is capable of detecting a single de novo mutation in ~5x107 sequenced wild-type bases.
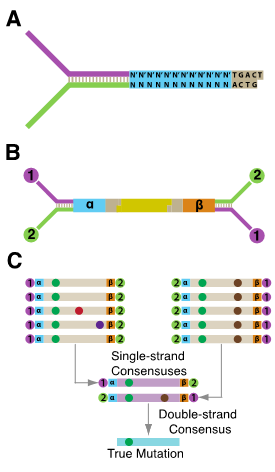
Briefly, sheared double-stranded DNA is ligated with a complementary double-stranded UMI (Fig. 2A). Following ligation, the individually labeled strands are amplified, resulting in many copies that share a common UMI derived from each single parental strand of DNA (Fig. 2B). After sequencing, reads sharing the same UMI are grouped together and a consensus sequence for each position in the read is calculated for each family to create a ‘single-strand consensus sequence’ (SSCS), with each SSCS being derived from an individual strand of DNA (Fig. 2C). The SSCS cannot filter out PCR errors arising during the first round of PCR, frequently induced by DNA damage. To remove these errors, the complementary UMI derived from the same duplex DNA among the SSCS reads are compared to each other. The base identity at each position in a read is kept in the final consensus if the two strands match perfectly at that position (Fig. 2C). Any deviations from the reference genome after mapping of the ‘duplex consensus sequence’ (DCS) reads are considered true mutations.
Getting Started
We require an initial consultation so that we may assist you with planning and design of your experiment. Please contact us to schedule a consultation. We will discuss your project goals, sample type, timeline, and estimated cost. The major determinants of overall project cost will be the number of desired target depth, region of interest number and size, available DNA amounts, and sequencing requirements.
NanoString GeoMx Digital Spatial Profiling
The NanoString GeoMx DSP permits direct quantification of proteins or RNA transcripts to spatially resolved anatomic structures in tissue sections. This service is in partnership with the Digital Spatial Profiling Core. We will work with you and the DSP Core to facilitate the use of departmental sequencers, as well as the initial computational processing of your data. Please refer to the DSP Core website for more information on this technology.
Questions?
Have other applications not listed here? Please contact Dr. Scott Kennedy to determine if we can help you make use of pre-existing analysis pipelines.